Trait Prediction (PHENOTREX)
Bacterial Trait Prediction with PICA
We use a reimplementation of the PICA software called phenotrex (Feldbauer et al. 2015, https://doi.org/10.1186/1471-2105-16-S14-S1). Phenotrex makes use of python3 and Linear SVM of scikit-learn to train models and perform predictions on sets of orthologous groups of proteins (ENOGs) from the eggNOG database. The reimplementation improved training and prediction of speed and reduced the memory footprint of models and virtual memory greatly.
Phenotrex creates binary vectors that reflect presence and absence patterns of orthologous groups for each sample. Those vectors are then used to create predictions for every model.
Phenotrex forms predictions based on the absence or presence of the potential to express proteins similar to those contained in orthologous groups within eggNOG, thus the quality of the input genomic material is directly correlated with the quality of predictions.
Since metagenomic bins are liable to be both contaminated with genetic material stemming from organisms other than the main OTU of the bin, and also potentially missing crucial genetic material required to perform the roles of the main OTU, information about bin quality must be integrated into the prediction to allow users to gauge overall prediction quality.
For this reason, Phenotrex output encompasses two quality measures which can be used to mask low quality predictions:
- Prediction confidence: The internal probability of class membership within the linear SVM model used by Phenotrex (provided via Platt scaling). Low prediction confidence is caused by the absence of ENOGs learned to be important for predicting the true class, or the presence of ENOGs learned to be important for predicting the false class. Prediction confidence can thus be taken to mean the confidence of the model in its classification under the assumption that the set of ENOGs presented to it is the true set of ENOGs present in the bin.
- Mean Balanced Accuracy: This is a set of values pre-computed on the model by sampling the training data to various different completeness and contamination levels, and assessing the resulting balanced accuracy (i.e., the mean of sensitivity and specificity when performing cross-validation) of the model. This measure is assigned to a prediction on a bin by retrieving the value computed with training data sampled to comparable completeness/contamination values. Low mean balanced accuracy at unfavorable completeness and contamination levels is due to the increased probability of ENOGs crucial for predicting the true class being absent, and ENOGs increasing the probability of the false class being present (due to contamination). Mean balanced accuracy can thus be taken to be the expected accuracy of the prediction given the bin’s completeness and contamination levels.
To establish the estimated mean balanced accuracy of a trained phenotrex model for different completeness and contamination levels of bins, a 5x cross-validation on the training data is performed while randomly removing ENOGs from the positive training instances (simulating varying levels of completeness) and/or randomly adding ENOGs from negative training instances (simulating varying levels of contamination).
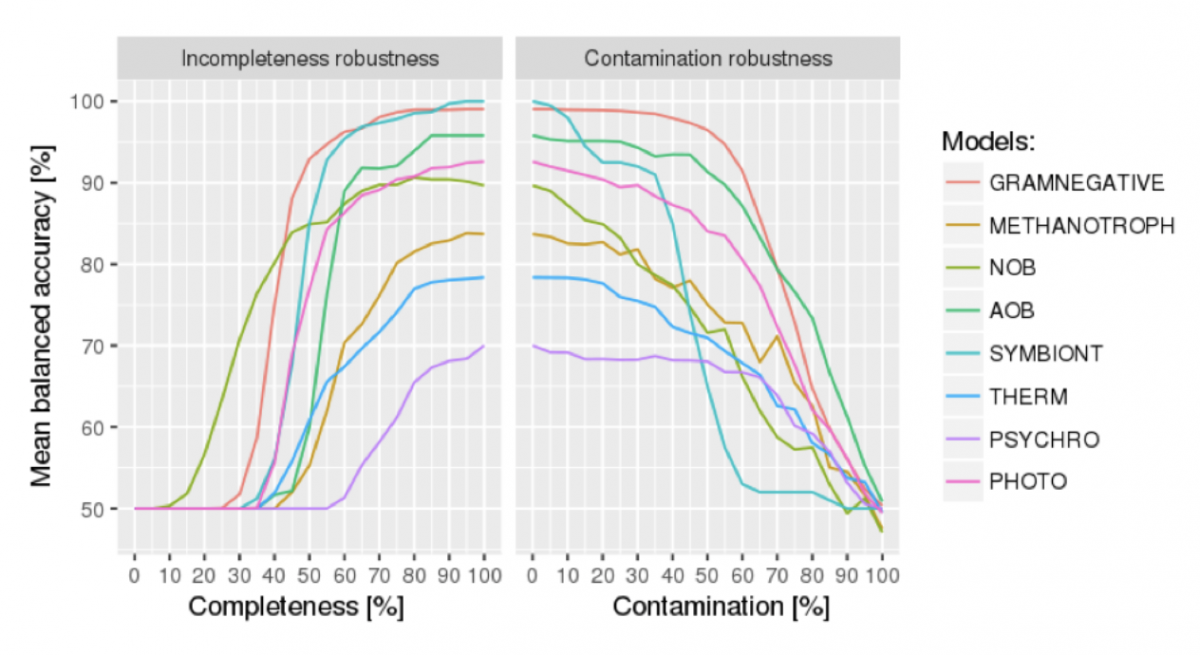
Figure 1, detailing the mean balanced accuracy of several PhenDB models at different simulated completeness and contamination levels of the training data.
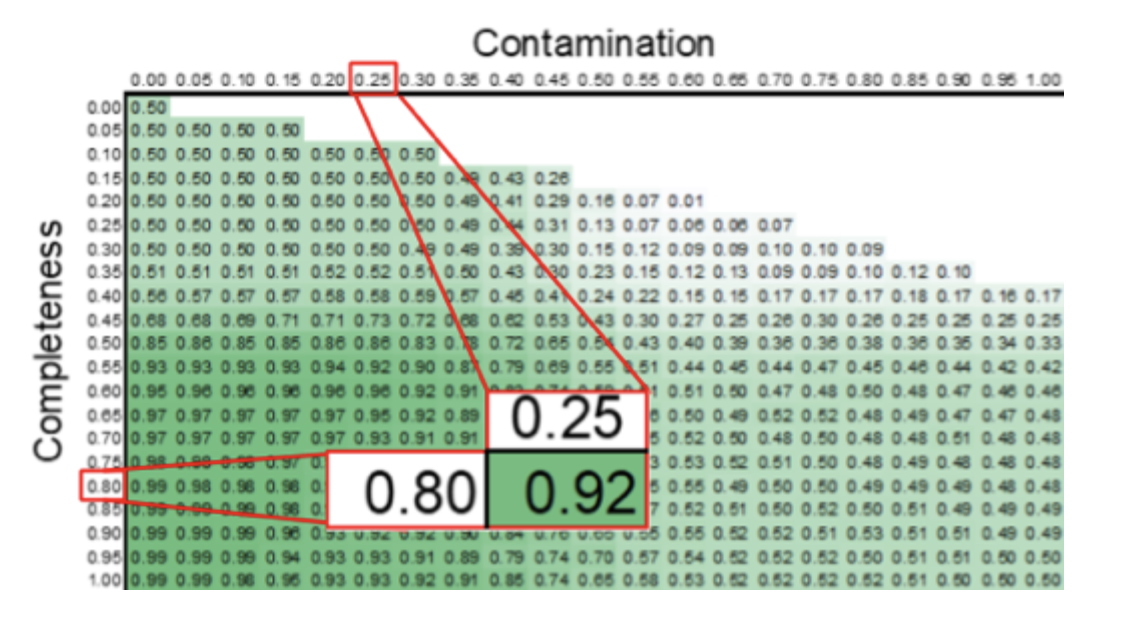
Figure 2, the finalized accuracy matrix of a PhenDB model which is used to retrieve the mean balanced accuracy of a newly uploaded bin for this model.
Training Data for PHENOTREX
As PhenDB relies on a machine learning-based technique (phenotrex) for trait predictions, a model for each trait is pre-trained on a dataset of genomes belonging to organisms with known traits. Each model description contains a letter identifying the source of its training data (in brackets below).
- The core (C) set of trait models consists of manually curated datasets for 9 traits that were present in the PICA publication of Feldbauer et al. (2015), as well as models for type III, IV and VI secretion systems and a model for determining archaea.
- Nitrogen-related models (Nitrogen fixation, Ammonia oxidation, Nitrite oxidation) and autotrophy models were manually curated with expert knowledge (E) of our colleagues in the Department of Microbiology and Ecosystem Science in Vienna (http://dmes.univie.ac.at/). (Acknowledge all who helped).
- Additional 18 models for metabolic products were extracted from existing metabolic models of the "Virtual Metabolic Human" project at https://www.vmh.life/
- Models for aerobe and anaerobe respiratory capabilities, spore formation, motility and Gram stain as well as a model for dissimilatory Sulfate reduction capabilities were trained on genome-trait pairs extracted from a large corpus of scientific literature, using a machine learning-based text mining approach (T). Thus, please note that, for this model type, mislabeled training instances (wrongly identified by text mining) may be present in the model’s training data set. However, due to the wider variety and greater quantity of training data, and the selection of training instances based on published phenotype alone, the predictive power of (T) models is usually significantly higher than (C)ore and especially Metabolic modeling models.
You can explore the training data for each model here: http://phendb.csb.univie.ac.at/browse-phendb. If you are missing specific traits, or are interested in collaboration, please let us know.
Release announcements
News

Latest publications
Contact
- +43 1 4277 76681
- +43 1 4277 876681
- contact.cube@univie.ac.at